About CARTaGENE PheWeb
This platform presents multiple analyses conducted using CARTaGENE data to the scientific community as well as the public at large. The CARTaGENE cohort is specific to Quebec and is a regional subset of the Canada-wide "Canadian Partnership for Tomorrow's Health"
This platform displays Phenome-wide association (PheWAS) results from the CARTaGENE cohort (i.e. statistical associations between individuals' trait information and genomic variations across the cohort).
Genotype Data
We built this version of PheWeb using N=29,337 CARTaGENE participants genotyped on five different arrays.
| Short name | Desription | N Individuals | N Variants |
|---|---|---|---|
| Archi | GSA v3 + Multi disease panel (MHI_GSAMD-24v3-0-EA_20034606_C1) | 1,909 | 688,795 |
| 17K | GSA v2 + Multi disease panel + addon (CaG_addon_v1_20037253_A2) | 17,286 | 645,075 |
| 4K | GSA v2 + Multi disease panel (GSAMD-24v2-0_20024620_A) | 4,179 | 728,919 |
| 5K | GSA v1 + Multi disease panel (GSAMD-24v1- 0_20011747_A1) | 5,237 | 658,296 |
| 760 | GSA v1 (GSA-24v1-0_A1) | 726 | 626,377 |
The genotyped CARTaGENE participants are from six Quebec regions: Gatineau, Montreal, Quebec, Saguenay, Sherbrooke, Trois-Rivieres. Only the 17K and 5K arrays included participants from all six regions.
| Region | N Individuals in 17K (%) | N Individuals in 4K (%) | N Individuals in 5K (%) | N Individuals in 760 (%) | N Individuals in Archi (%) |
|---|---|---|---|---|---|
| Gatineau | 757 (4.4) | None | 326 (6.2) | 1 (0.1) | None |
| Montreal | 12,354 (71.5) | 2,921 (69.9) | 3,423 (65.4) | 522 (71.9) | 1,302 (68.2) |
| Quebec | 2,428 (14.0) | 877 (21.0) | 715 (13.7) | 61 (8.4) | 410 (21.5) |
| Saguenay | 556 (3.2) | 151 (3.6) | 228 (4.4) | 94 (12.9) | 196 (10.3) |
| Sherbrooke | 698 (4.0) | 229 (5.5) | 359 (6.9) | 48 (6.6) | None |
| Trois-Rivieres | 493 (2.9) | None | 186 (3.6) | None | None |
Only the 17K and 5K arrays included many participants recruited in both phase 1 and phase 2 of the study.
| Phase | N Individuals in 17K (%) | N Individuals in 4K (%) | N Individuals in 5K (%) | N Individuals in 760 (%) | N Individuals in Archi (%) |
|---|---|---|---|---|---|
| 1 | 9,828 (56.9) | 4,177 (100.0) | 2,503 (47.8) | 715 (98.5) | 1,908 (100.0) |
| 2 | 7,456 (43.1) | 1 (0.0) | 2,734 (52.2) | 11 (1.5) | None |
Pre-imputation
We started with the genotype data on genome build GRCh37/hg19 saved in five different files (i.e. one per array in PLINK format). We confirmed that each array's genotype data had undergone the initial QC at CARTaGENE.
- Variants showing departures from Hardy-Weinberg Equilibrium (P-value < 10e-6) were removed.
- Variants with call rate <95% were removed.
- Samples with call rate <95% were removed.
- Samples with discordance between reported and inferred sex were removed.
Our steps for CARTaGENE genotype data imputation to the TOPMed reference panel included:
- Lift over each array's genotype data to human genome build GRCh38.
- Combine genotype data from the five arrays and keep only shared variants.
- Impute combined genotype data.
The following sections describe each of these steps in detail.
Lift over to GRCh38
We converted each array's genotype data to genome build GRCh38 and aligned it to the "+" strand. Our conversion included the following steps:
- We lifted genetic positions from GRCh37 to GRCh38 using UCSC LiftOver tool and the corresponding chain files.
- We excluded variants that did not map to GRCh38, mapped to alternate contigs in GRCh38, or located on the mitochondrial chromosome.
- We aligned A1/A2 alleles in the original PLINK files to the REF/ALT alleles on the "+" strand using the plink2reference.py script.
- We excluded palindromic variants (A/T and G/C), variants for which A1/A2 alleles didn't match REF/ALT, and for which A1/A2 alleles were not A, C, G, or T.
- We renamed all variants to follow the CHR:POS:REF:ALT notation.
- If multiple variants had the same name, we kept only one with the highest call rate using the plink_dupvar_prioritize.py script. We observed such duplicated variants only on customized genotyping arrays.
- All heterozygous genotypes for males in chromosome X and Y non-PAR regions were set to missing.
The final number of variants after these steps was:
| Array | N Variants in GRCh37 | N Variants in GRCh38 |
|---|---|---|
| Archi | 688,795 | 620,623 |
| 17K | 645,075 | 612,224 |
| 4K | 728,919 | 648,511 |
| 5K | 658,296 | 638,099 |
| 760 | 626,377 | 581,440 |
Merging array data
We merged the genotypes from the five arrays in the following steps:
- We looked for the individuals with the same ID across two or more arrays. We identified 3 individuals with same ID present on the 760 and 5K arrays. We kept only genotypes from the 5K array for these individuals due to higher array coverage.
- We merged arrays' genotypes and kept 481,223 shared variants (i.e. genotyped in all five arrays), Y chromosome specific variants were not considered.
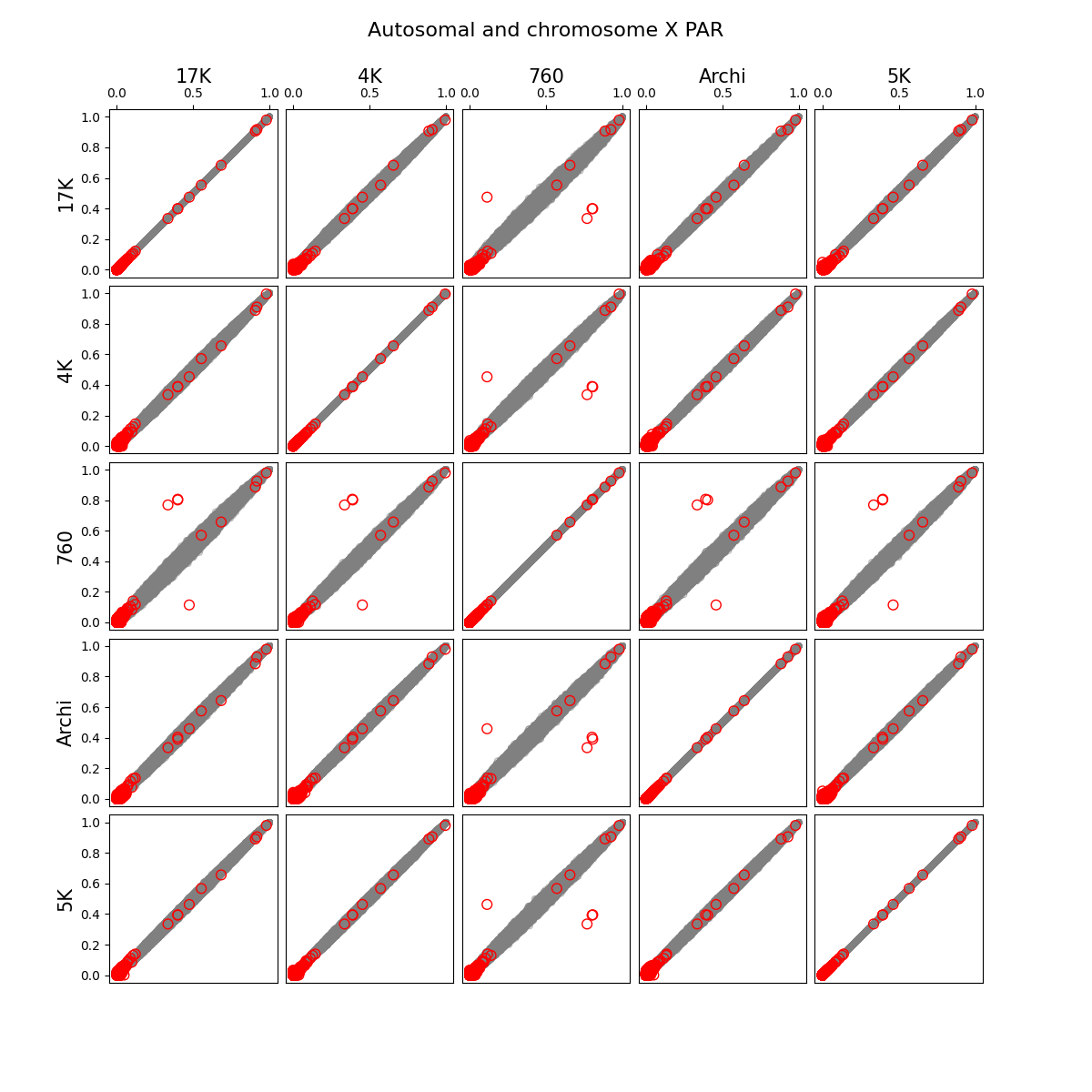
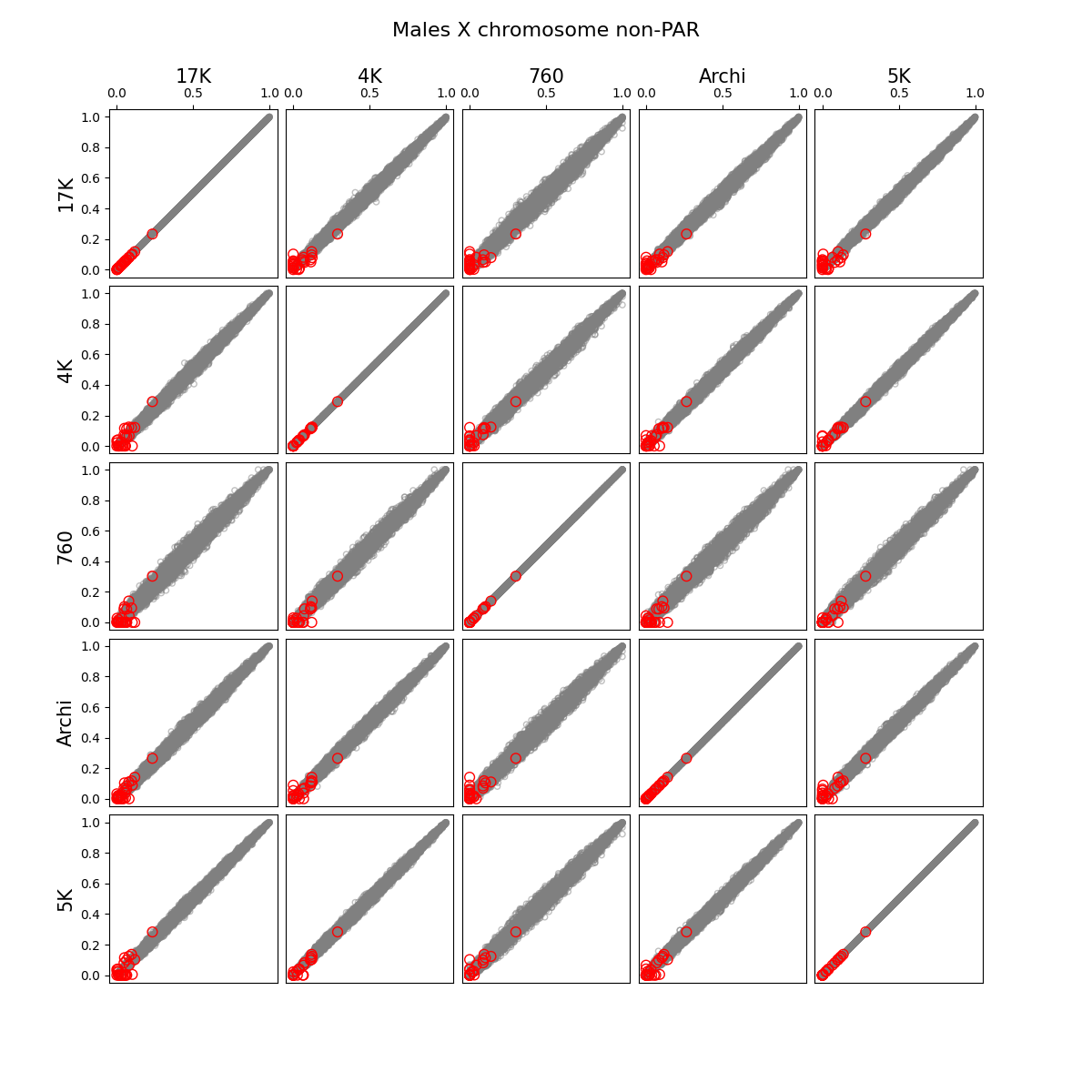
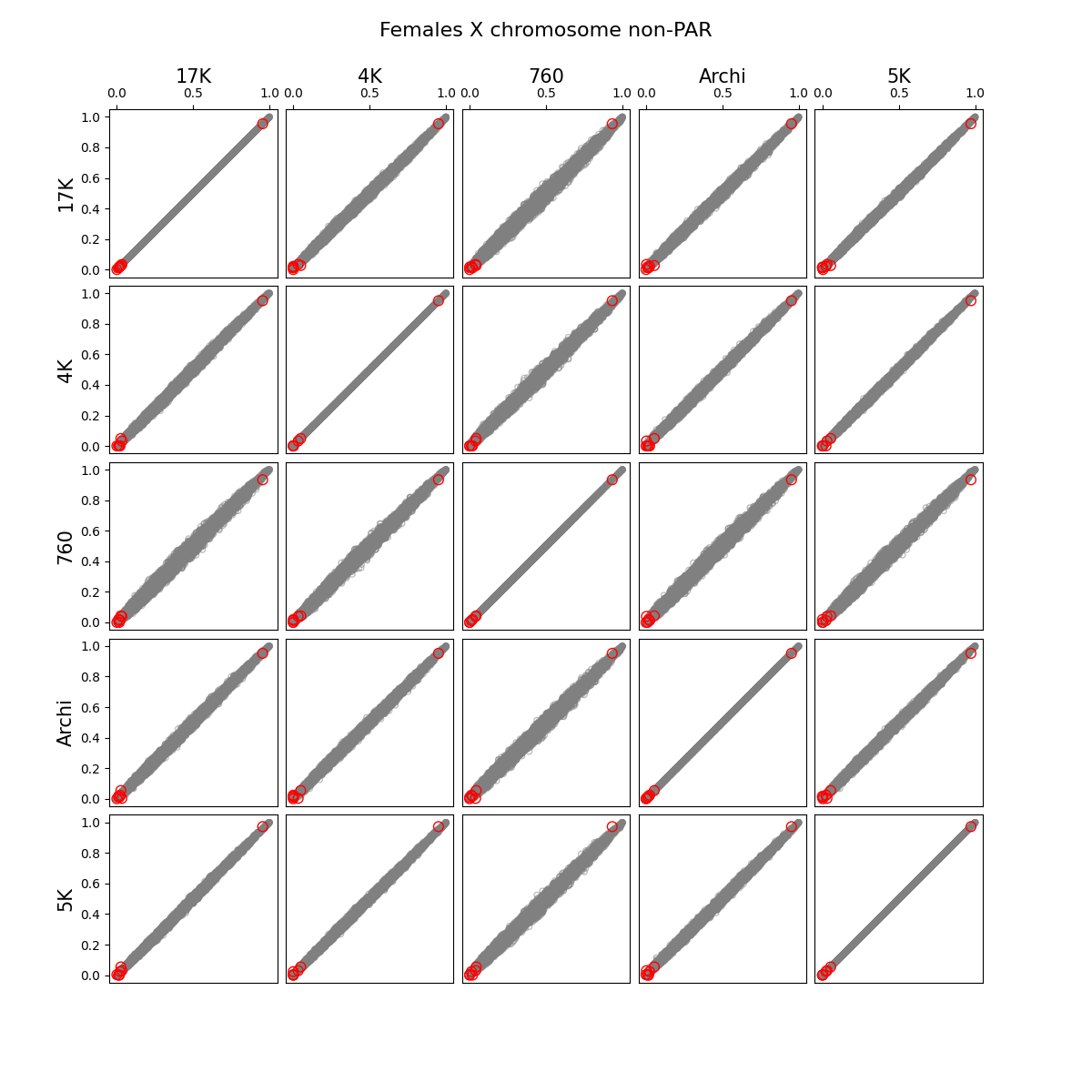
- We identified and removed 1,315 variants (Figure 1) which showed statistically significant deviations of allele frequencies between arrays while accounting for genetic ancestry:
- We projected all individuals to the 1000 Genomes Project reference panel's PCA space using TRACE.
- We removed all related individuals with 3rd, 2nd, and 1st degree of relatedness (PLINK's --king-cutoff option set at 0.0442).
- We performed likelihood ratio test comparing two linear regression models
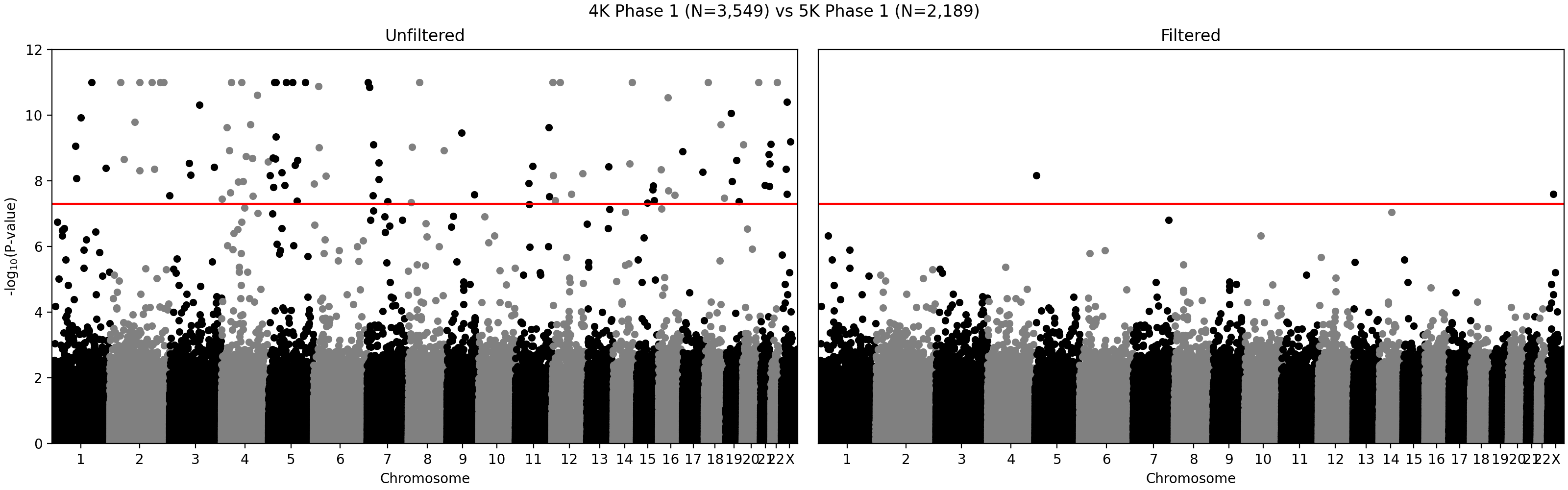
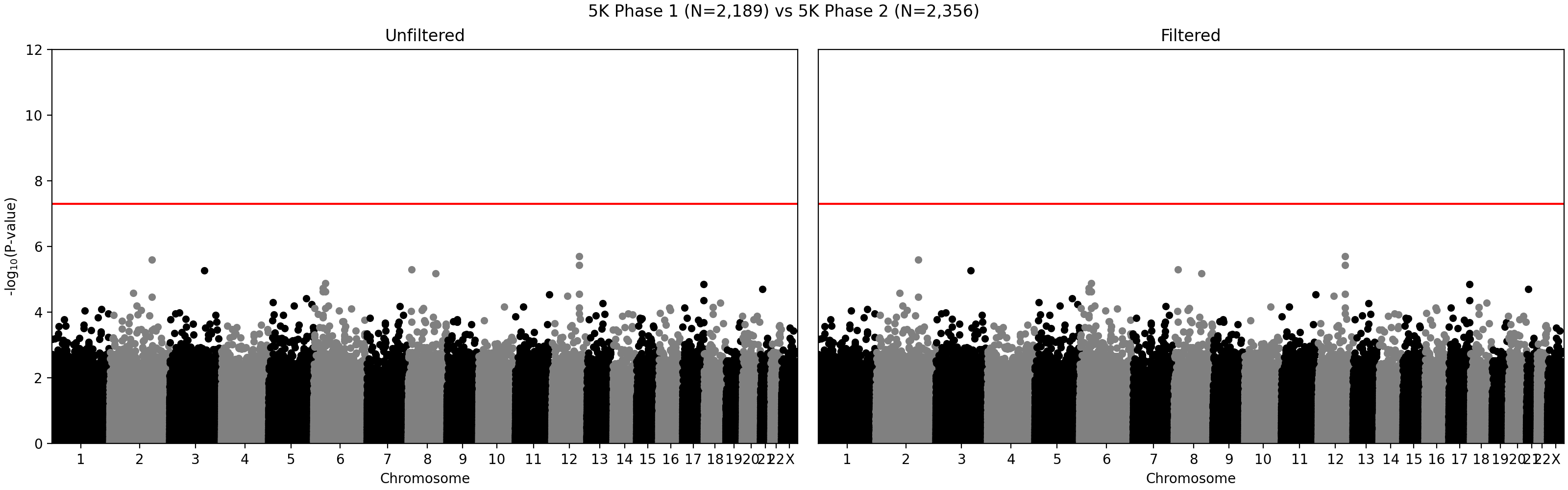
G ~ m + PC1 + ... + PC4 + A2 + ... + A5andG ~ m + PC1 + ... + PC4, where G - individual genotype (0, 1, or 2), m - intercept corresponding to the average alternate allele frequency, PCi - i-th principal component, Aj - indicator variable for array j. Assuming random sampling and no batch effects, the array variable should not have impact on predicting individual-specific allele frequency. The test is implemented in the compare_ancestry_adjusted_af.py script. For the non pseudo-autosomal region of the X chromosome, we performed this test for both sexes separately. - We ran Firth association test (PLINK's --glm firth option) using unrelated individuals and genotyping array and phase as outcomes, and sex and first five PCs as covariates. We confirmed that our filtering removes variants with potential batch effects (Figure 2). In addition, we didn't see any evidence of systematic batch effects between phase 1 and 2 (Figure 3).
The final merged dataset included 29,330 individuals and 479,908 variants.
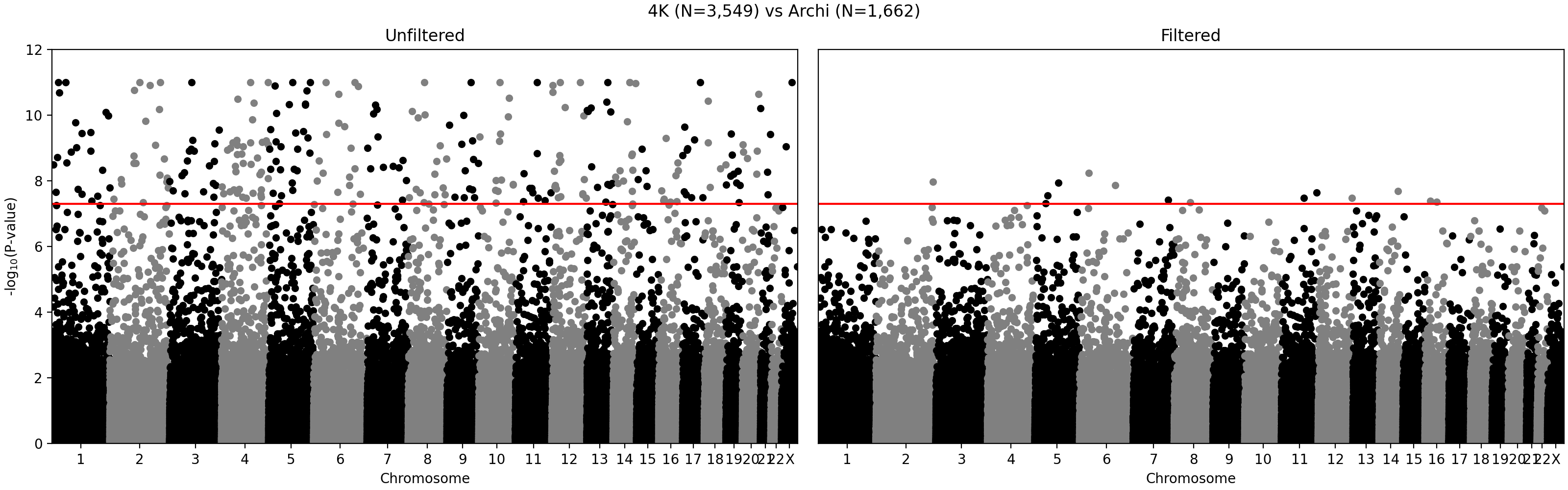
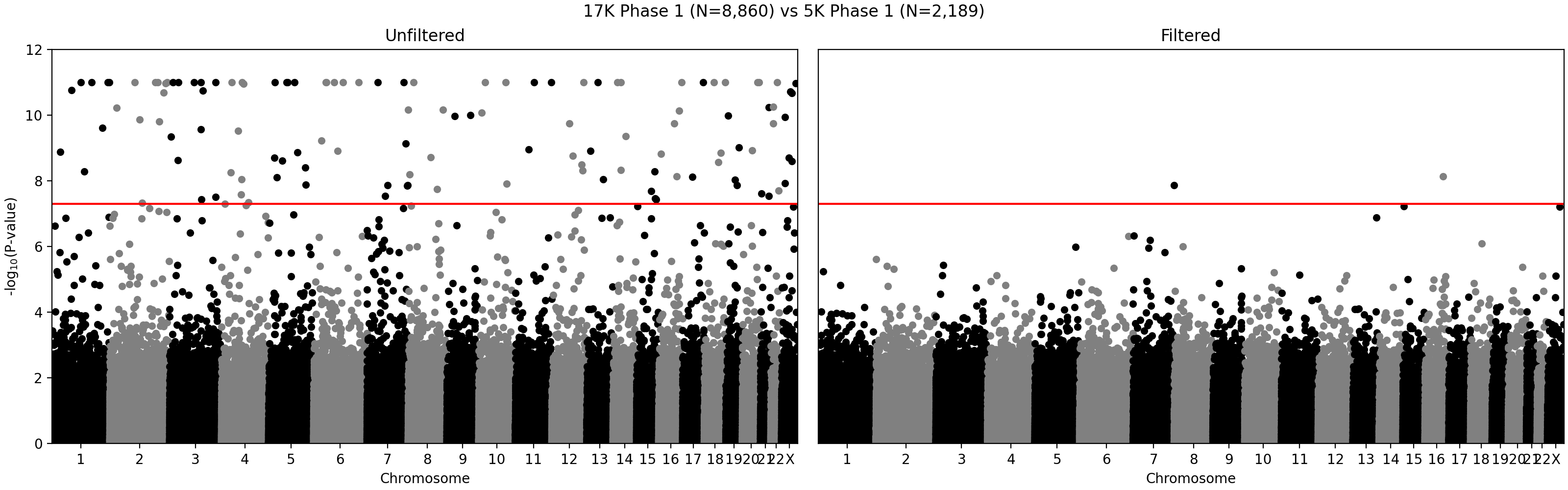
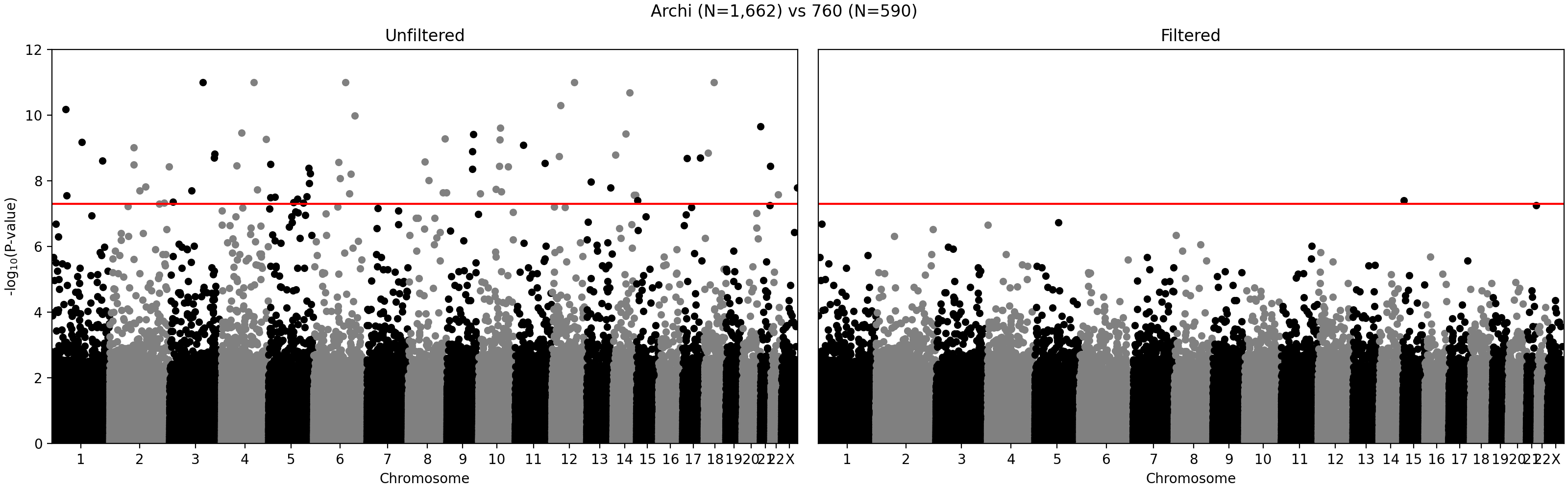
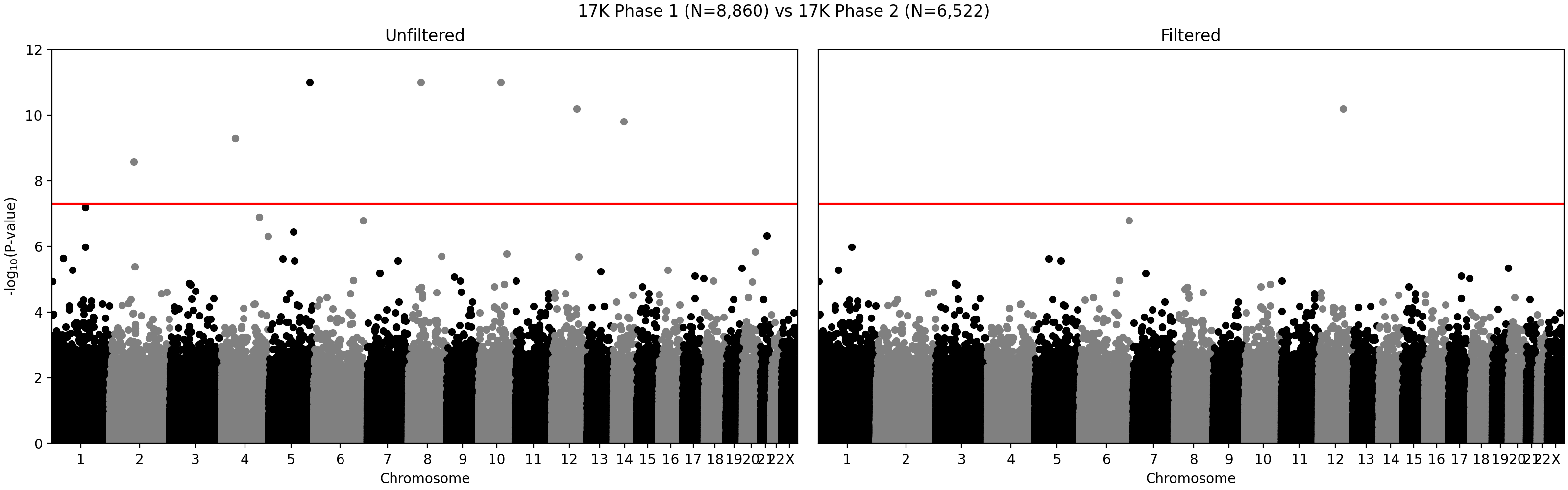
Check against the TOPMed reference panel
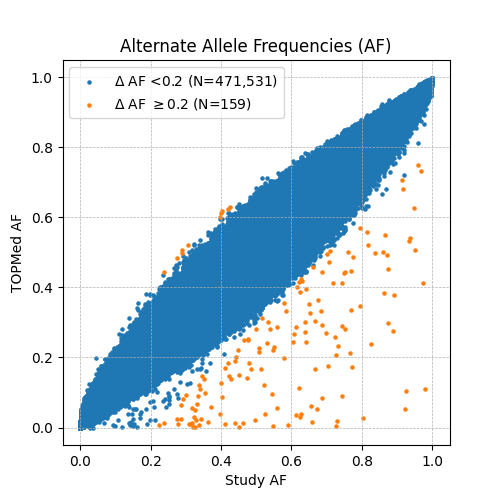
We removed a further 159 variants which showed substantial differences in alternate allele frequencies when compared to the TOPMed reference panel.
- We downloaded TOPMed Freeze 8 allele frequencies from the BRAVO variant browser.
- We identified and removed variants from CARTaGENE's merged array data with AF difference >0.2 compared to TOPMed Freeze 8 (Figure 1). See plink_freq_vs_topmed.py script for more details.
Imputation to the TOPMed reference panel
We imputed CARTaGENE genotypes using the TOPMed reference panel through the TOPMed Imputation Server at the NHLBI BioData Catalyst platform. At the moment of our analyses, the TOPMed Imputation Server was limiting the imputation jobs to 25,000 individuals. To overcome this limitation, we split the CARTaGENE imputation into two batches. The following sections describe our approach.
Splitting array data for imputation
For imputation purposes, we split CARTaGENE's merged array data into two overlapping batches (Figure 1):
- We randomly permuted the list of 29,330 individual IDs.
- We generated two separate lists: the 1st included the top 25,000 IDs, and the 2nd had the bottom 25,000 IDs from the permuted list. Thus, the overlap between the two lists was 20,670 individuals.
- We generated two sets of VCFs for imputation - each included only the individuals from the corresponding lists.
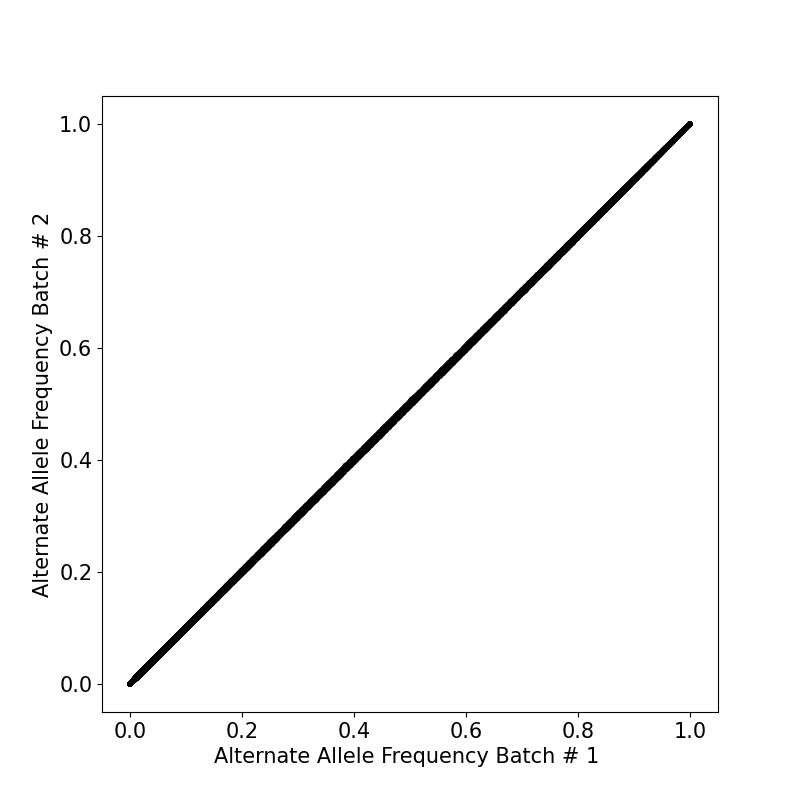
By generating the overlap between two batches, we ended up with similar population allele frequencies between variants in two sets (Figure 2).
Imputation results
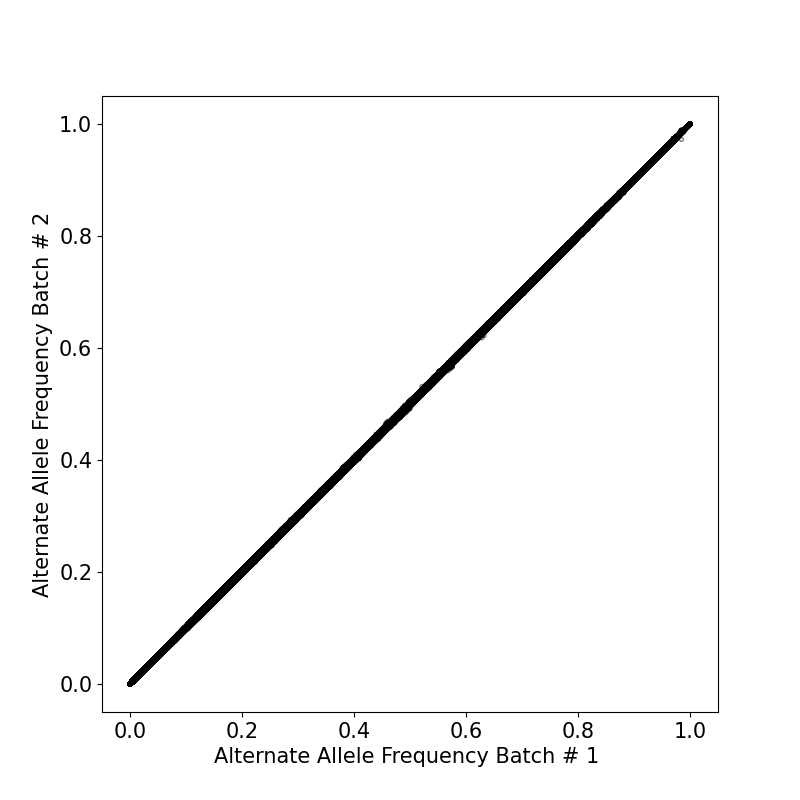
We imputed the 307,841,049 variants in each of the two batches. The alternate allele frequencies (AF) of imputed variants were similar in both batches (Figure 1).
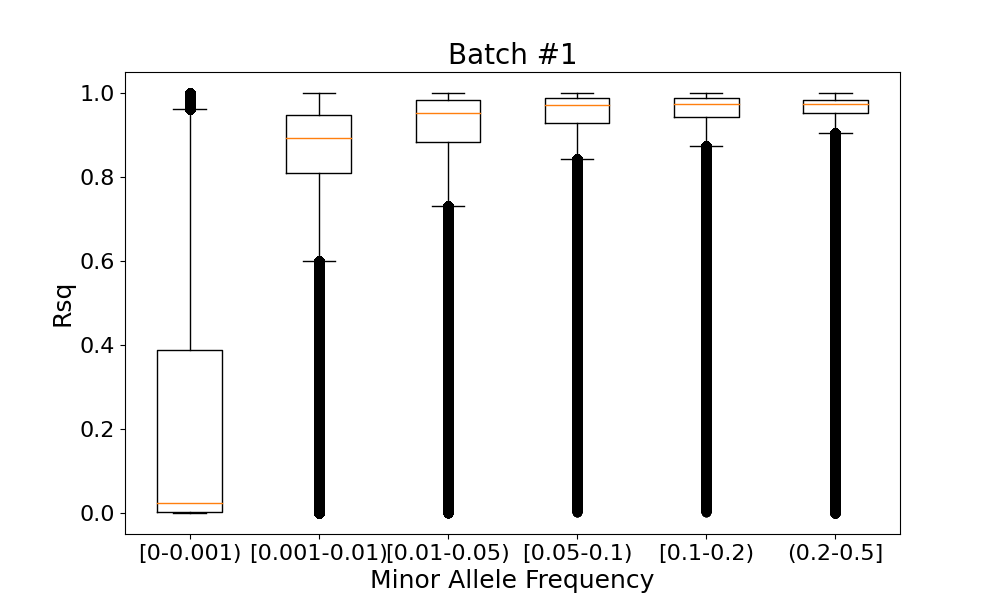
The low-frequency variants with MAF<0.001 had the lowest imputation qualities (median Rsqbatch #1=0.02401 and median Rsqbatch #2=0.02409), while more frequent variants with MAF>0.01 had the highest imputation qualities (median Rsqbatch #1>0.93842 and median Rsqbatch #2>0.93825) (Figure 2).
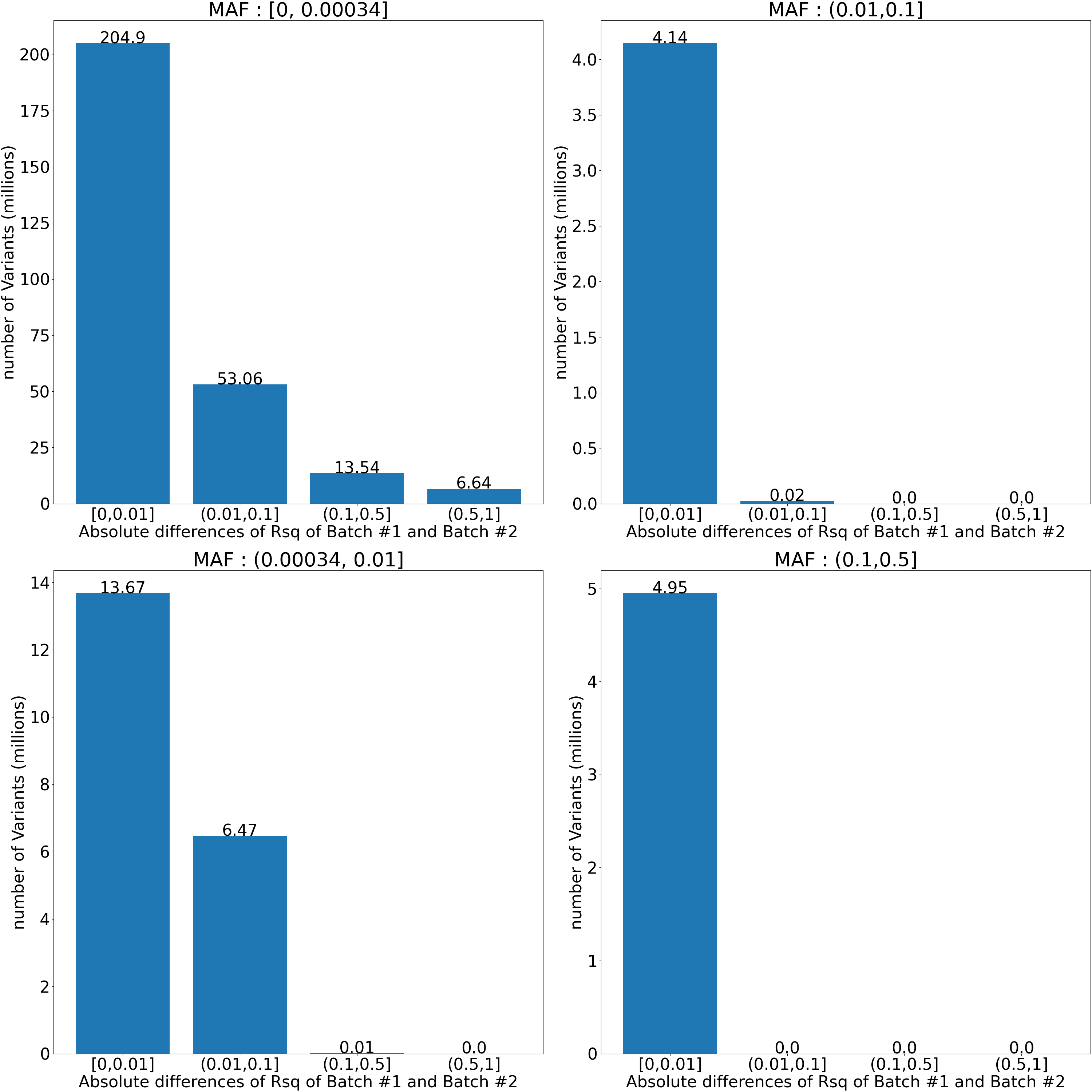
For each imputed variant in two batches, we looked at the absolute difference between its imputation qualities Rsqbatch #1 and Rsqbatch #2 in these batches (Figure 4). We observed the largest Rsq difference (>0.5) only in variants with the lowest minor allele frequency (MAF<0.00034095, corresponding to 20 alternate alleles). For the vast majority (>99%) of variants with MAF>0.01, the Rsq difference didn't exceed 0.01.
Merging imputation results
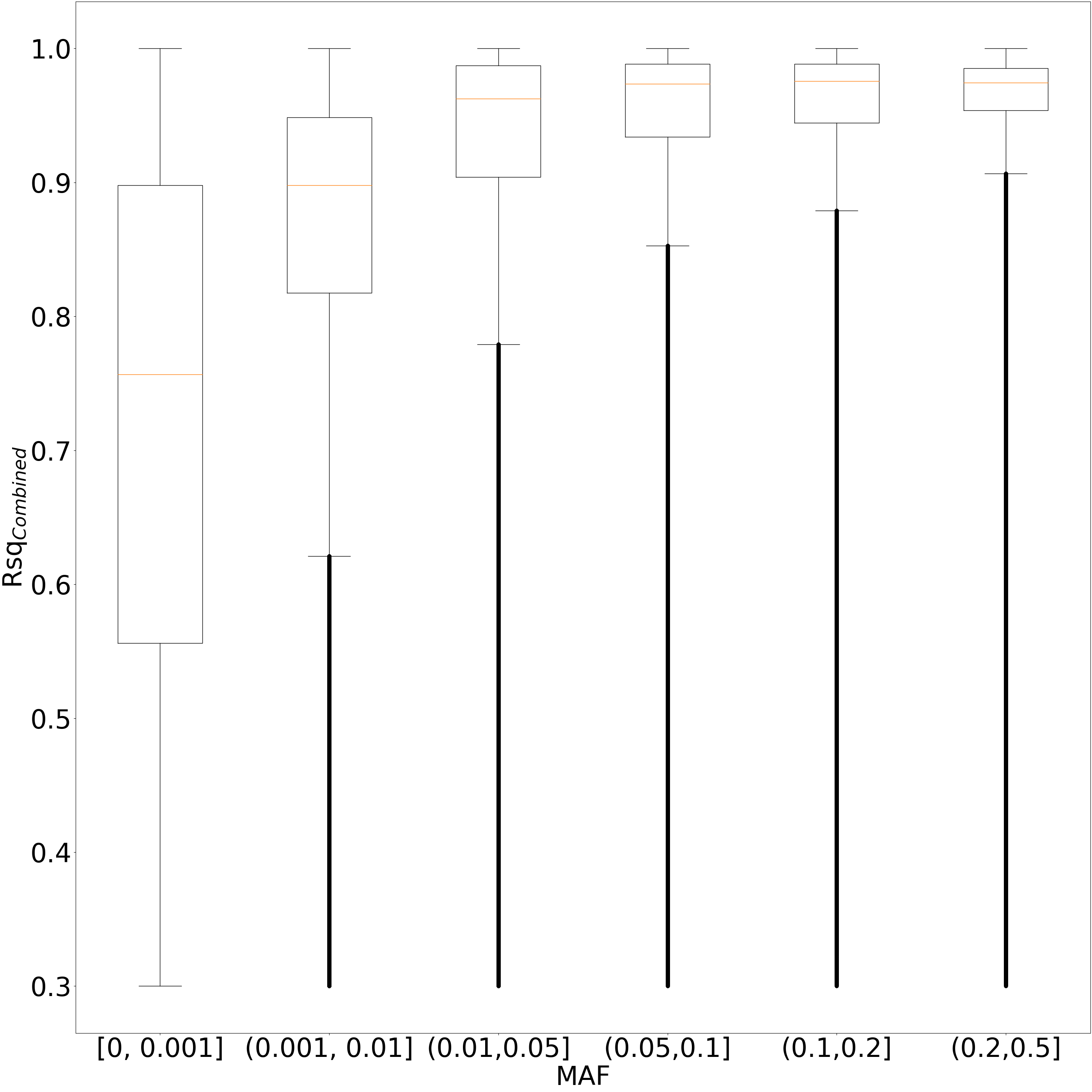
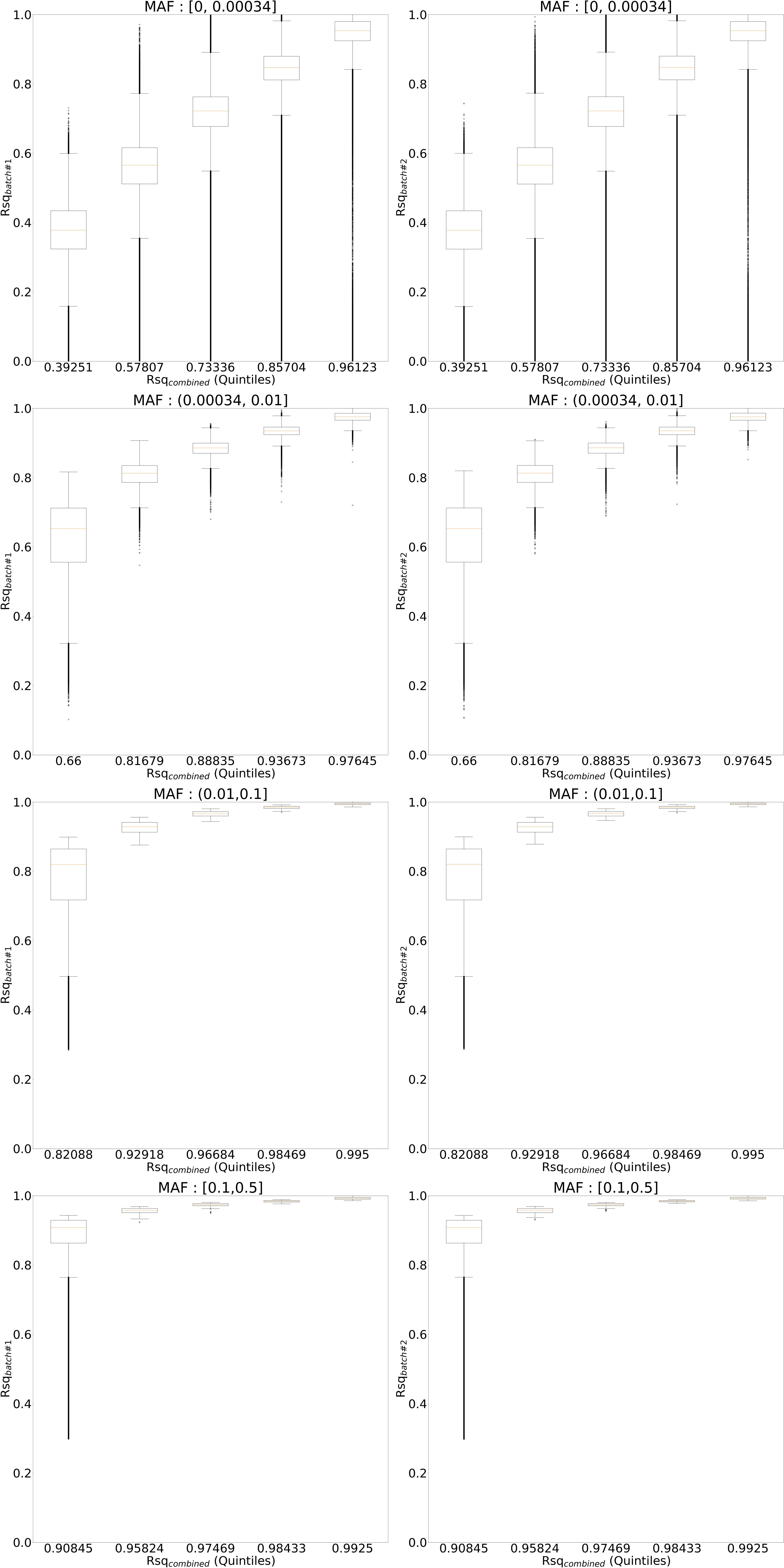
We merged the imputed genotypes from two batches using the merge_minimac_dosages.py script. For the dosages imputed in both batches, we selected the dosage from the batch with the highest imputation quality (Minimac Rsq) for the corresponding variant. After, we filtered out variants with the combined imputation quality Rsqcombined≤0.3 (Figure 1). The underlying distributions of Rsqbatch #1 and Rsqbatch #2 were similar for each quantile of Rsqcombined (Figures 2).
The final dataset included 103,574,557 imputed variants (Table I).
| MAFcombined | [0,0.00034] | (0.00034,0.01] | (0.01,0.1] | (0.1,0.5] |
|---|---|---|---|---|
| Number of variants | 74,532,694 | 19,663,301 | 4,244,017 | 5,134,545 |
Phenotype Association
In this version of the CARTaGENE PheWeb, we restricted our genome-wide association analyses (GWASs) to individuals of European genetic ancestry. We used Regenie to run GWASs. The following sections describe the selection of individuals, phenotypes, and the corresponding Regenie arguments.
Selecting individuals of European genetic ancestry
We selected CARTaGENE participants of inferred European genetic ancestry using the following steps:
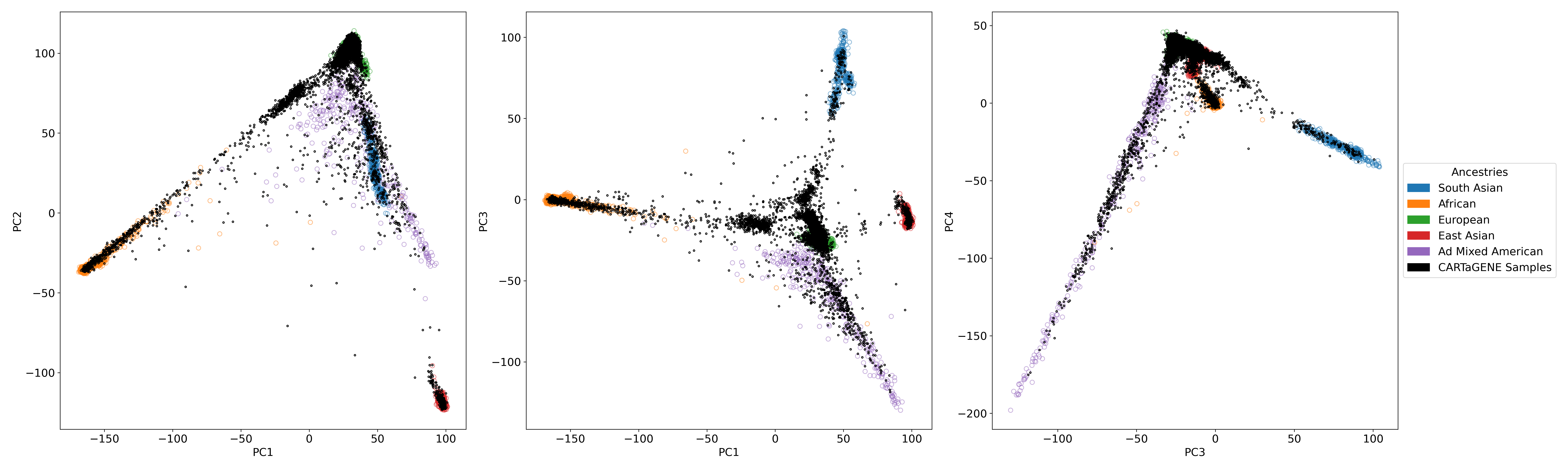
- We projected CARTaGENE samples to the reference PCA space constructed using unrelated individuals in the 1000 Genomes Project (1000G) (Figure 1).
- For each CARTaGENE individual, we computed the Mahalanobis distance to each of the 5 superpopulations in the 1000G PCA space (Admixed American, African, East Asian, South Asian, European). We used the first 6 principal components and Mahalanobis.py to compute the Mahalanobis distance.
- We assigned a CARTaGENE individual to a superpopulation if the corresponding Mahalanobis distance's P-value was ≥0.001 and all other P-values were <0.001 (Table 1).
- We removed 879 CARTaGENE individuals for whom the inferred genetic ancestry didn't match the ETHNICITY variable value derived from the questionnaire data.
- We confirmed that 25,017 CARTaGENE individuals with assigned European genetic ancestry: (a) cluster together with European ancestry individuals in 1000G (Figure 2), (b) have a vast proportion of European ancestry autosomal chromosomes (Figure 3).
| Genetic ancestry | Ad Mixed American | African | East Asian | South Asian | European | Other |
|---|---|---|---|---|---|---|
| Number of individuals | 479 | 460 | 225 | 124 | 25,896 | 2,146 |


Kinship and Cryptic Relatedness
We analyzed other structural aspects potentially present within the data :
- We estimated the relationship between individuals of all ancestry using the King software.
- 4 duplicate pairs were identified from inconsitency between DNA inference and questionnaire (i.e Are you a twin ? Do the pair share the same birthday?). The sample with the sample with the higher variant call rate within each pair was kept. Thus, our final European dataset is comprised of 25013 individuals.
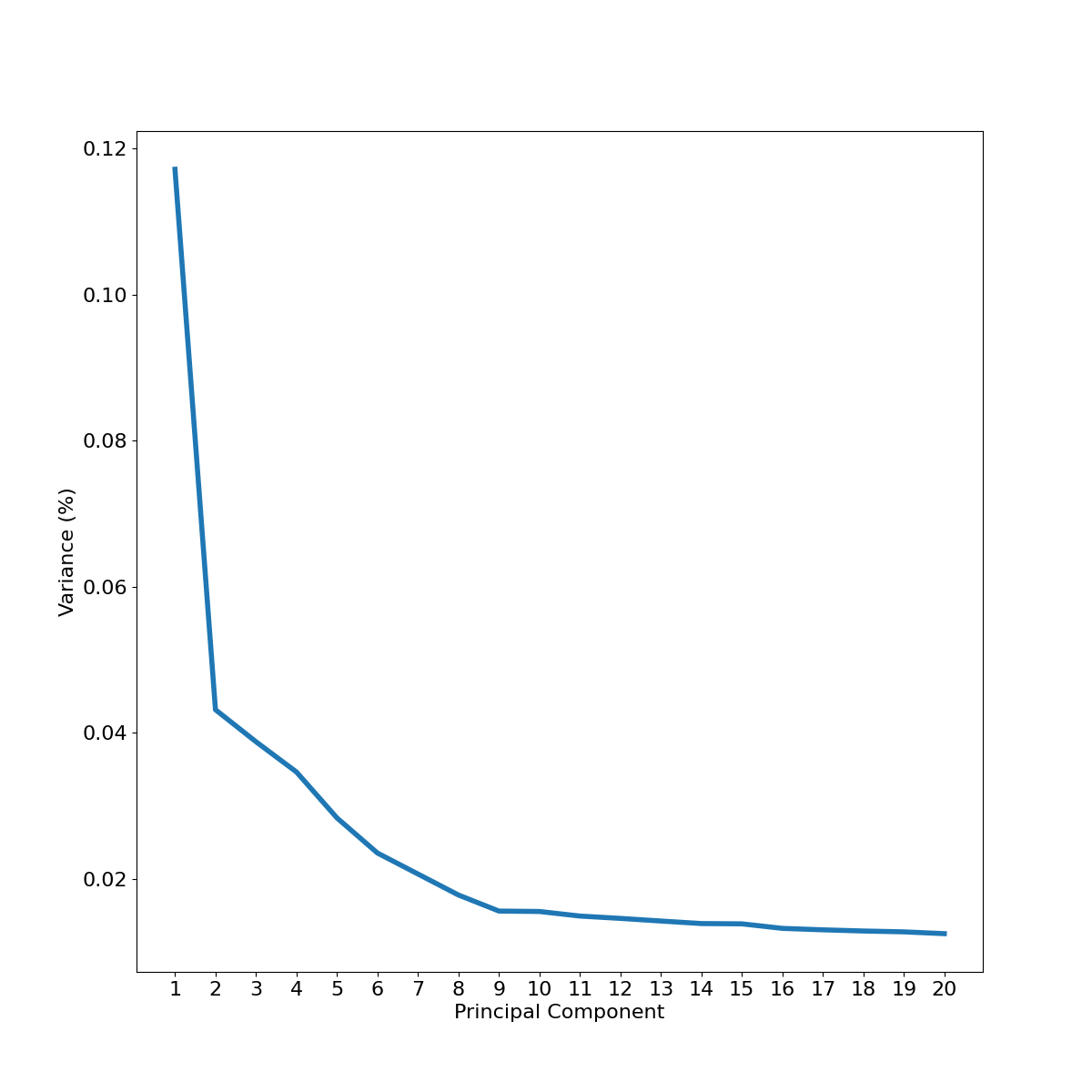
- We further explored any structure within the European samples using a PCA Analysis.
| Relationship | Non duplicate Monozygotic Twin | Parents / Offsprings | First Siblings | Second Degree | Third Degree |
|---|---|---|---|---|---|
| Number of individuals | 4 | 101 | 1,157 | 1,221 | 7,205 |
Phenotypes
We used binary and continuous phenotypes from the baseline assessment. To select the phenotypes of potential interest, we used the meta-information for CARTaGENE variables in the COMBINED_CATALOG_v2_9_7 - OCT2021 catalogue. We based our phenotype selection on the sample size estimated using a subset of CARTaGENE individuals of European genetic ancestry.
We selected 326 binary phenotypes to perform GWASs in the following way:
- We generated a list of binary phenotypes and the corresponding sample sizes using the prepare_binary_variables.py script. For each phenotype, this script performs the following:
- checks if the variable is coded as described in the meta-information file;
- recodes "NO" and "YES" to 0 and 1;
- recodes everything that is not 0 or 1 to "NA";
- counts the total number of non-missing values and number of cases and controls (stratified by sex).
- We manually curated the resulting list and removed phenotypes that:
- had a small sample size (N<1000)
- had a low number of cases (Ncases<10)
- were not relevant (e.g. LANGUAGE_SPOKEN_...)
We selected 375 continuous phenotypes to perform GWASs in the following way:
- We generated a list of continuous phenotypes and the corresponding sample sizes using the prepare_continuous_variables.py script. For each phenotype, this script performs the following:
- checks if all values are numeric;
- recursively recodes missing values to “NA”
- checks at least 10 unique numeric values are present
- counts the total number of non-missing values and number of unique values (stratified by sex)
- computes mininum, maximum, mode, mean, and median (stratified by sex)
- We manually curated the resulting list and removed phenotypes that:
- had a small sample size (N<100)
- were not relevant (e.g. LAST_MEAL_WHEN_HOUR_24)
- had single observation for each value except mode
Association model
We performed GWASs on >700 phenotypes using Regenie (version 3.0.1.) software, which handles unbalanced case-control ratios and accounts for population structure and relatedness. Our automated Nextflow pipeline for executing Regenie in a highly parallel way is available here.
In Regenie's Step 1 (whole genome model), we used the merged array genotype data with the following filters:
- MAF ≥0.01
- genotype missigness per variant <0.01
- genotype missigness per sample <0.01
- HWE P-value ≥1e-15
- removed low-complexity and high LD regions
- pruned variants in high LD (r2>0.9)
In Regenie's Step 2 (single-variant association testing), we used the imputed data and the following settings:
- Covariates: genotyping array (categorical), sex, age, age2, 1-10 PCs
- Rank inverse normal transformation for continuous phenotypes
- Firth approximation when p-value <0.05 in binary phenotypes
PheWeb
The CARTaGENE PheWeb uses the open-source PheWeb framework. The customized source code for this instance is available here.
Acknowledgements
- Authors : Vincent Chapdelaine, Sarah Gagliano Taliun and Daniel Taliun
- All computational analyses were performed using compute resources of Digital Research Alliance of Canada, and the Digital Research Alliance of Canada hosts this site.
- We thank all CARTaGENE participants who volunteered their time and information for research.